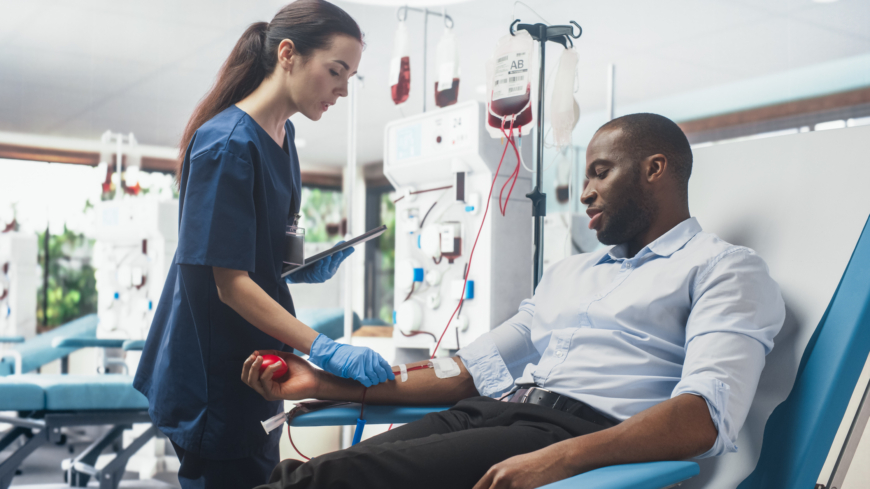
Thalassæmi er en arvelig blodsygdom, der forårsager mangel på hæmoglobin – det iltbindende protein, der findes i røde blodlegemer. Det er et kollektivt navn for flere forskellige varianter af tilstanden, med den fællesnævner, at det kan føre til anæmi, blodmangel. Thalassæmi kan i dag behandles med stamcelletransplantationer eller symptomatisk med blodtransfusioner samt et bestemt kemoterapeutisk lægemiddel. Nylige videnskabelige fremskridt er genterapi, der i fremtiden kan hjælpe patienter, der ikke har en matchet, nært beslægtet donor til rådighed. Medianalderen for dødsfald ved thalassæmi er næsten 30 år lavere end for den almene befolkning
Thalassæmi er en sjælden diagnose
I Norden er thalassæmi en sjælden tilstand, men der er ingen pålidelige data om, hvor mange mennesker der har diagnosen. Der er sandsynligvis omkring 100 personer, der har den alvorlige form for beta-thalassæmi, som kræver blodtransfusioner, og denne tilstand forekommer næsten udelukkende hos mennesker med genetisk oprindelse fra bestemte områder i verden. I Norden er der omkring 250 mennesker, der har en moderat til svær form for beta-thalassæmi.
Globalt er tilstanden en af de mest almindelige genetiske sygdomme, og mere end 150 millioner mennesker bærer gener, der kan forårsage thalassæmi. Tilstanden er mest almindelig i de geografiske områder af verden, hvor malaria forekommer, eller i områder, hvor malaria tidligere var almindelig, dvs. omkring Middelhavet, i Mellemøsten, Indien, Sydøstasien, Kina og Afrika.
Genetik årsagen til forskellige former for thalassæmi
Thalassæmi er forårsaget af skade på et af de gener, der producerer hæmoglobin. Der er mange gener, der kan forårsage thalassæmier, og det er meget individuelt, hvor meget hæmoglobin kroppen kan producere hos forskellige personer med tilstanden.
Hæmoglobin kan se forskelligt ud hos forskellige personer, men består normalt af to forskellige typer proteinkæder, beta (ß) og alfa (a). Hvor meget der produceres af disse to proteinkæder, afhænger af personens arveanlæg. Ved thalassæmi er de gener, der koder for disse proteiner, påvirket. Tilstanden er opdelt i alfa- og beta-thalassæmier, afhængigt af hvilket af disse gener der påvirkes.
Beta-thalassæmi
Ved beta-thalassæmi er genet for betakæden muteret, og dermed er produktionen af betakæden nedsat. Beta-thalassæmi har forskellige sværhedsgrader afhængigt af, om personen kun er bærer af ét fejlkodet gen (thalassæmi minor), eller om begge gener er fejlkodet (thalassæmi major)
De personer, der har beta-thalassæmi minor, er raske, og tilstanden indebærer normalt ikke nogen større fysiske eller psykiske problemer. Der kræves heller ingen særlig behandling. Derimod er det vigtigt for en bærer at spise en varieret vitamin- og jernrig kost.
Thalassæmi major forårsager mere betydelige problemer, og diagnosen stilles normalt i de første leveår på grund af alvorlig anæmi (blodmangel) og mobilitetsproblemer. Personer med thalassæmi major har behov for transfusioner.
Alfa-thalassæmi
Alfa-thalassæmi er forårsaget af forandringer i alfa-kæden. Alfakæden findes i to gener på hvert kromosom, hvilket vil sige i fire forskellige gener, og derfor findes sygdommen i fire forskellige varianter:
Alfa-thalassæmi major. Her er alle fire alfagener forandret. Dette fører til så dårlig ilttransport, at fostre med denne mutation allerede dør i livmoderen.
Alfa-thalassæmi minor. Her er to af alfakæderne påvirket af genforandringer. Denne version af sygdommen forårsager mild til moderat anæmi, men kan også være mere alvorlig i nogle tilfælde.
Hæmoglobin H-sygdom. Denne variant af alfa-thalassæmi har tre berørte alfakæder. Hæmoglobin H forårsager alvorlig anæmi, der kan blive endnu værre ved feber, indtag af visse lægemidler og eksponering for visse smitstoffer. Sygdommen kræver ofte også behandling med blodtransfusioner.
Stille bærer af alfa-thalassæmi. Her er kun ét gen påvirket, og ofte kan denne variant ikke engang ses i almindelige blodprøver. Det forårsager heller ikke nogen signifikante symptomer. Bærere af alfa-thalassæmi har ofte røde blodlegemer, der er lidt mindre end normalt. Den eneste måde at stille diagnosen med sikkerhed på er gennem DNA-analyse.
Arvelighed ved thalassæmi
Sygdomsgenerne ved thalassæmi arves på en autosomal recessiv måde. Det betyder, at en person kun bliver syg, hvis han eller hun modtager sygdomsgenet fra begge forældre. Selvom begge forældre har sygdomsgenet, er de selv raske, fordi det andet gen i genparret fungerer normalt. Disse forældre har 25 procent sandsynlighed for at få børn med alvorlig sygdom.
Symptomerne på thalassæmi varierer
Symptomerne varierer afhængigt af hvilken type thalassæmi, det drejer sig om. Personer med en moderat eller svær form kan have mange forskellige symptomer ud over blodmangel, såsom modtagelighed over for infektioner, nedsat kondition, knogleskørhed og hjertesvigt. Lever og milt kan blive forstørret, og man kan få diabetes. Nogle børn med thalassæmi vokser langsomt og bliver lavere end forventet. Disse børn kan også få vækstændringer i knoglestrukturen i ansigt og kranie. Nogle får aldrig symptomer, mens andre kan få symptomer på anæmi, såsom træthed, nedsat kondition eller bleghed. Symptomerne vil være mest fremtrædende i forbindelse med infektioner eller kraftig fysisk anstrengelse.
Patienter med svær thalassæmi udvikler symptomer i omkring tre til seks måneders alderen. De berørte børn bliver blege, sløve og vokser dårligt. I de alvorligste tilfælde dør barnet allerede i fosterstadiet.
De fleste symptomer, der forårsages af anæmi, kan undgås med regelmæssige blodtransfusioner, men det er en metode, der påvirker livskvaliteten for patienten og de pårørende negativt.
Diagnosticering af thalassæmi
I lande med struktureret thalassæmipleje udføres der screening af forældre, der er i risiko for at bære sygdommen. I Danmark er der et målrettet screeningsprogram for familier og mennesker af genetisk oprindelse fra områder, hvor thalassæmi er almindeligt. Dette sikrer tidlig og korrekt diagnose gennem screening af nyfødte børn efter fødslen, som derefter modtager tilstrækkelig pleje på specialiserede centre.
Der skal tages blodprøver for at kunne stille diagnosen. Oftest ses først anæmi, som sammen med oplysninger om, at personerne kommer fra områder med høj forekomst af thalassæmi, giver en mistanke om thalassæmi. Når mere almindelige årsager til anæmi, såsom jernmangel, er blevet udelukket, kan man gå videre med prøver, der analyserer de forskellige dele af hæmoglobinproteinet, og en DNA-analyse. Det giver en indikation af hvilken type thalassæmi, det drejer sig om.
For personer med milde former for thalassæmi bliver diagnosen måske aldrig stillet. Derfor kan det for kvinder under en graviditet, hvor kroppen har et øget behov for at kunne danne nye røde blodlegemer, være første gang der udvikles anæmi som følge af sygdommen. Hvis udredningen derefter viser thalassæmi, tages der normalt også prøver fra den kommende far for at se, om han ligeledes er bærer af sygdomsgener. Formålet med dette er at se, om der er risiko for, at det ventede barn kan have en alvorlig form for thalassæmi.
Thalassæmi – lindrende, men ikke helbredende behandling
Folk, der ikke har symptomer på grund af deres thalassæmi, behøver heller ikke nogen behandling eller opfølgning. De personer, der har milde symptomer, kan i nogle tilfælde have brug for blodtransfusion, for eksempel i forbindelse med infektioner eller mere alvorlige betændelsestilstande. Personer med alfa-thalassæmi kan have brug for tilskud af folinsyre for at kompensere for folinsyremangel.
For de fleste mennesker er der ingen behandling, der kan helbrede thalassæmi. Ved svær thalassæmi er hyppige, regelmæssige blodtransfusioner derfor afgørende hele livet. De mest alvorligt syge patienter kan have brug for en transfusion hver tredje uge, mens andre kan klare sig med længere intervaller. I gennemsnit er der behov for 17 blodtransfusioner om året. Transfusionerne kan dog betyde, at kroppen har et overskud af jern, som den ikke kan udskille, hvilket kan føre til skade på forskellige organer. Derfor kan lægemiddelbehandling med jernbindende stoffer være nødvendig for at reducere jernabsorptionen i organerne.
Før der blev indført blodtransfusionsterapi og lægemidler til reduktion af jernoverskud, overlevede personer med thalassæmi sjældent i mere end ti til femten år. For patienter med thalassæmi major og transfusionsafhængig thalassæmi er den forventede levetid stadig omkring 30 år kortere end normalt, omkring 45-50 år.
Stamcelletransplantation og genterapi
Stamcelletransplantation er en behandling, der kan lindre sygdommen, men metodens brug er begrænset på grund af de risici og komplikationer, der er forbundet med transplantationen, samt mangel på donorer i familien, som har de rette gener.
Personer med thalassæmi har defekte blodlegemer på grund af afvigelser i deres blodstamceller. Med en stamcelletransplantation kan der produceres sunde blodlegemer fra nye stamceller. Ved thalassæmi kommer stamcellerne normalt fra en anden person. For at udføre en sådan transplantation skal vævstyperne hos modtageren og donoren være så ens som muligt. Vævstypen bestemmes af et protein kaldet HLA, og der er en stor mangfoldighed af forskellige HLA-varianter. Grunden til, at det er vigtigt at finde et så tæt match som muligt, er, at modtagerens eget HLA-system kan genkende fremmede celler og begynde at angribe dem.
Fordi der er så mange forskellige HLA-varianter, kan det være svært at finde en passende donor til transplantation. Søskende har 25 procent chance for at være et match, fordi de arver deres gener fra to forældre, og hver forælder har to sæt gener. Det betyder, at der er fire mulige kombinationer af gener, der kan overføres til søskende.
Genterapi er den seneste videnskabelige udvikling. Det har givet mulighed for genetisk modifikation af de gener, der er involveret i hæmoglobinproduktionen. Dette har ført til en løbende udvikling af genterapi til β-thalassæmi og kan vise sig at være et alternativ for patienter, der ikke har en matchet, nært beslægtet donor til rådighed i fremtiden.
Livet med thalassæmi
Det er en daglig kamp at leve med transfusionsafhængig beta-thalassæmi (TDT). For at overleve er personer med denne genetiske blodsygdom afhængige af regelmæssige blodtransfusioner, hvilket fører til en ophobning af overskydende jern i kroppen. Dette kan føre til:
- organskader
- organsvigt
- andre komplikationer med hjerte, lever og nyrer
- øget risiko for knogleskørhed, diabetes og problemer med adenohypofysefunktionen.
Som et resultat af stærkt reduceret (eller mangel på) hæmoglobin påvirkes antallet og funktionen af røde blodlegemer. Dette giver symptomer som:
- anæmi/blodmangel
- træthed
- åndenød
- smerter
Hos spædbørn kan det føre til problemer som vanskeligheder med at falde til ro, gulsot og problemer med at indtage føde.
Den smerte og træthed, man kan føle i hele kroppen, når man lider af thalassæmi, påvirker naturligvis den daglige livskvalitet ekstremt negativt. Ud over symptomerne på sygdommen er håndteringen af TDT krævende og tager meget tid. Patienterne bruger mange timer hver måned på lægebesøg, som ofte kræver støtte fra familie og plejepersonale. I gennemsnit kræves 17 transfusioner om året. I en undersøgelse rapporterede patienter, at de i gennemsnit brugte næsten 16 timer hver måned på at håndtere deres sygdom, og at de følte sig “fastlåst” af sundhedssystemet på grund af håndteringen af sygdommen.
Håndteringen af sygdommen kan gøre det svært at have et fuldtidsjob, det gør det svært at tilbringe tid sammen med familie og venner, og det er ofte meget mentalt belastende. Det er konstateret, at af de personer, der har TDT, lider 20 % af depression og 40 % af angst. Dette til sammenligning med den generelle befolkning, hvor 3,4 % anslås at have depression og 3,8 % en angstlidelse. På trods af fremskridt i behandlingen har personer med TDT typisk en kortere levetid end den generelle befolkning med en medianalder for dødelighed på 45 år i Storbritannien og 37 år i Frankrig.
På trods af disse udfordringer findes der ny genterapi og stamcelletransplantation, der har til formål at reducere behovet for transfusion, men tilgængeligheden af og tilskuddet til dette varierer fra land til land. Det er vigtigt at øge bevidstheden om denne sygdom og fortsætte med at søge efter bedre behandlingsmuligheder for at forbedre livskvaliteten for de berørte.